

第一作者:Jianchao Dong
通讯作者:Xuyong Yang & Ning Wang
通讯单位:上海大学,吉林大学
DOI:https://doi.org/10.1038/s41565-024-01852-6
研究背景
尽管在绿色和红色金属卤化物钙钛矿发光二极管(PeLEDs)方面取得了重大进展,但蓝色PeLEDs,尤其是深蓝色,由于严重的相分离导致的电致发光光谱偏移以及在宽带隙钙钛矿发射极中低激子利用率,仍然大大落后。
本文亮点
本文提出了一种多价固定策略,通过引入一种含氟氧多原子分子来实现高效且光谱稳定的深蓝色PeLEDs。系统的实验和广泛的5000fs从头分子动力学模拟揭示,多价效应的关键在于氢键(F···H–N)、离子键(F–Pb)和配位键(C=O:Pb)与钙钛矿的三种相互作用,它们协同稳定钙钛矿相并增强激子的辐射复合。深蓝色钙钛矿发射器的激子浓度和激子复合率分别增加了1.66倍和1.64倍。本文的目标PeLEDs在459nm的深蓝色发射波长下显示出高达15.36%的峰值外量子效率,并且在0.45 mA cm−2的恒定电流密度下寿命达144分钟。此外,深蓝色PeLEDs在稳定的驱动电流下保持了恒定的光谱峰值,其CIE色品坐标为(0.136, 0.051),持续时间为60分钟。
图文解析
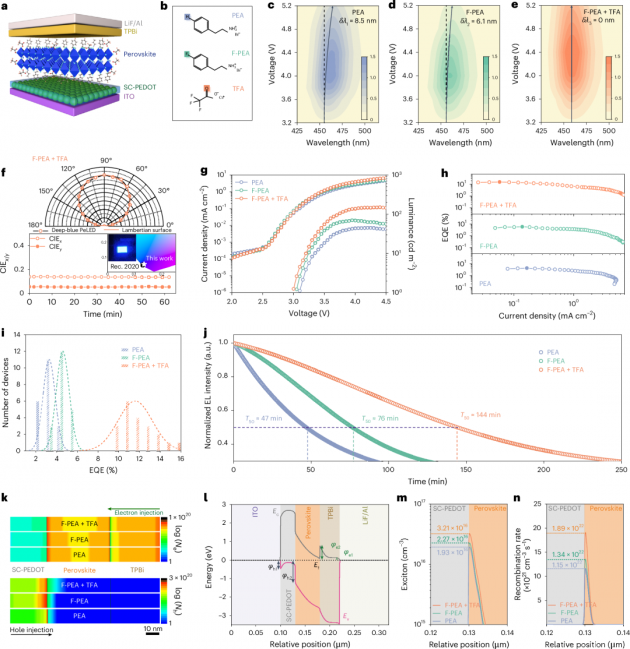
图1| 深蓝色PeLEDs的器件性能和自洽光电模拟
要点:
1.本文制造了深蓝色PeLEDs,其器件结构如下:涂有氧化铟锡(ITO)的玻璃基底/掺杂柠檬酸钠的聚(3,4-乙撑二氧噻吩)聚苯乙烯磺酸盐(SC-PEDOT)作为空穴传输层,该层可以同时提高PEDOT:PSS层的空穴注入能力和形貌,如本研究团队之前的工作所报道的那样(图1a)。RDP发光层通过旋涂前驱体溶液并使用抗溶剂(间二甲苯)辅助的方法沉积。图1b展示了三种有机分子的结构,分别基于(苯乙基溴化铵)PEA、(4-氟苯乙基溴化铵)F-PEA和(铯三氟乙酸盐)TFA。所有器件均展现出深蓝色电致发光(EL)。然而,在电压从3.2V增加到5.2V时,PEA处理的器件中观察到EL红移8.5纳米(图1c),而F-PEA处理的器件也显示出轻微的峰值偏移(6.1纳米)(图1d)。相比之下,F-PEA+TFA处理的器件完全没有EL峰值偏移(图1e),且峰值EL亮度高于PEA处理和F-PEA处理的器件。
2.F-PEA+TFA处理的PeLED的角发射强度遵循朗伯分布(图1f),并且CIE值可以在连续工作条件下保持不变。图1f的插图展示了一个在3.7V电压下工作的PeLED的生动照片,它发出明亮的深蓝色光,CIE色坐标为(0.136, 0.051),与Rec. 2020规定的深蓝色标准(0.131, 0.046)非常匹配。目标最佳性能的PeLED在459纳米处显示出高达15.36%的峰值外量子效率(EQE)(图1g,h),这在深蓝色PeLED领域大大超过了以往的工作。EQE再现性的PeLEDs与TFA也呈现在直方图中(图1i),平均EQE为11.56±1.7%,是PEA处理器件的三倍。
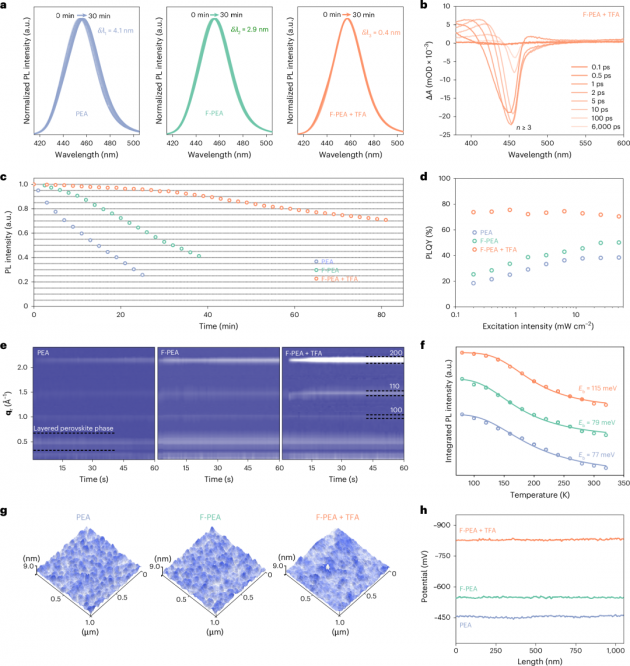
图2| RDP膜的光学和电学特性
要点:
1.本文评估了RDP薄膜的发射稳定性,以探索其在电致发光(EL)光谱特性上的改进。在80℃的加热条件下,本文记录了RDP薄膜的光致发光(PL)峰位(图2a)和PL强度(图2c)。观察到用PEA处理的RDP薄膜遭受严重的热猝灭和光学迁移。然而,用F-PEA处理的RDP薄膜显示出改善的稳定性,而F-PEA+TFA处理的RDP即使连续加热1小时后,仍保持其初始发射强度的80%以上。这是PEA处理的RDP保持其初始发射强度时间的十倍。为了深入了解RDP薄膜的量子阱宽度分布和光学性质,进行了瞬态吸收光谱分析。PEA和F-PEA样品表现出多n相分布,包括n=2的钙钛矿相。相比之下,在引入TFA后,F-PEA+TFA样品中的n=2区域明显受到抑制(图2b)。F-PEA+TFA样品的长组分1,792 ps表明非辐射复合路径减少,激发载流子被有效利用。
2.本文进一步通过稳态PL和紫外-可见吸收光谱分析了发光特性。发光强度的增加表明F-PEA和TFA分子对RDP薄膜具有优异的钝化效果。更高的PLQYs和更长的PL寿命也证实了缺陷密度的降低。此外,F-PEA+TFA处理薄膜的PLQYs显示出较弱的激发强度依赖性(图2d),表明其光学特性优越。而且,从PEA处理到F-PEA处理再到F-PEA+TFA处理的RDP样品,激子结合能(Eb)从77 meV增加到115 meV(图2f),光学声子能量(ћω)从47.52 meV增强到79.35 meV。这些结果表明,F-PEA+TFA处理的RDP经历了强烈的限制效应和热抗猝灭效应,这与PLQYs和热稳定性测量结果一致。有趣的是,钙钛矿的晶粒尺寸没有明显变化,而RDP薄膜的形态在加热前后有所不同。晶体在PEA衍生的RDP薄膜表面沉淀,而在F-PEA衍生的RDP薄膜上形成孔洞,表明加热时有机胺的损失。相比之下,在F-PEA+TFA衍生的RDP薄膜中可以清楚地看到晶粒的显著融合,同时基本上保持了薄膜的完整性。本文推测TFA与RDPs之间存在多价效应(多个化学键),导致有机阳离子紧密结合在钙钛矿表面上,增加了激子结合以及提高了钙钛矿结构的稳定性。
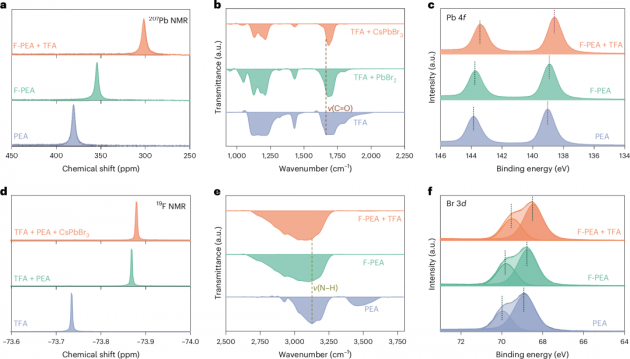
图3| 有机物与RDPs之间的多价效应
要点:
1.为了研究RDPs中的多价效应,本文进行了核磁共振(NMR)实验,以检测有机物与RDPs之间的强相互作用。对于F-PEA+TFA处理的RDP样品,207Pb的化学位移位于301.7 ppm,明显偏离了F-PEA处理(354.4 ppm)和PEA处理(380.7 ppm)的样品(图3a)。本文将这种偏移归因于F和Pb之间形成的离子键(F–Pb),或者羰基(C=O)中氧原子上的孤电子对向Pb2+离子的空6p轨道捐赠。进行了反射微傅里叶变换红外(FTIR)光谱和X射线光电子能谱(XPS)实验,以观察C=O伸缩振动(图3b)和Pb 4f的信号(图3c)。在TFA粉末的FTIR光谱中,来自C=O伸缩振动(ν(C=O))的峰位于1665 cm−1,在与CsPbBr3粉末混合后,波数移至更高的1682 cm−1,表明TFA与Pb2+阳离子之间的配位。
2.与PEA处理的RDP相比,F-PEA处理和F-PEA+TFA处理的RDP显示出Pb 4f核心能级的峰向较低的结合能偏移,进一步验证了配位键(C=O:Pb)的形成。从PEA处理的RDP到F-PEA+TFA处理的RDP,Pb 4f7/2的光电子发射能移(0.4 eV)从139.0 eV移至138.6 eV,与F-PEA处理的RDP相比,表明C=O:Pb的相互作用占主导。此外,由于C=O:Pb的配位键,Br 3d和Cl 2p(图3f)也显示出向较低结合能的偏移,导致Pb2+与卤素阴离子之间的静电相互作用发生变化。这些结果提供了证据,表明TFA钝化有效减少了由离子空位引起的非辐射复合。

图4| 通过多价效应固定深蓝色钙钛矿发射体的机制
要点:
1.本文使用密度泛函理论(DFT)计算进一步阐明了多价效应导致的固定机制。计算结果证实,Pb离子与周围的F和O离子有明显的电荷转移。通过DFT模拟引入晶体轨道哈密顿布居数(COHP)来量化和比较TFA和RDP之间的不同作用力(图4a)。观察到的O和Pb之间的最大作用力表明其主导相互作用,其次是F和Pb,以及F和H。这些结果与本文实验发现一致。有无有机组分的差异电荷密度分布图和电子定域函数清楚地展示了TFA分子与RDPs之间的相互作用。
2.本文对三种结构进行静电势表征,以揭示电荷分布并确定可能的相互作用。计算显示,氟与其周围的氢原子之间有明显的电荷转移(图4b),这是由于氢键的形成。因此,有机组分的空位形成能增加(图4c,d),说明了氟元素对胺的固定作用。此外,卤素离子的空位形成能也增加了,这与飞行时间二次离子质谱(TOF-SIMS)的结果一致。通过从头算分子动力学模拟对RDP样品的动态特性进行了表征。如图4e所示,在没有TFA的情况下,有机组分(乙胺(EA)分子)随时间逐渐从RDP的无机本体中脱离。然而,在TFA处理的RDP情况下,有机组成在模拟过程中直到5000 fs都保持不变。这一重要发现与RDP薄膜的稳定性测试结果一致(图2a)。
总结与展望
本文证明了使用多价效应策略可以实现来自RDPs的高性能深蓝色电致发光(EL),其峰值EQE在459 nm处达到15.36%,并且在稳态电场下具有显著的光谱稳定性。EL性能的提升归功于三种化学键提供的多重保护,这些化学键同时防止了相分离和离子迁移,并增加了激子的辐射复合。本文的实验结果、理论计算和半导体自洽光电模拟阐明了通过多价效应固定可以全面控制深蓝色RDPs的结构、晶体学和光电子性能。这一方法展示了RDPs实现高效且稳定的深蓝色PeLEDs的潜力。除了深蓝色发射外,所提出的策略很可能广泛适用于稳定其他颜色钙钛矿的晶体结构,为未来全彩显示技术的发展提供支持。
原文详情:Dong, J., Zhao, B., Ji, H. et al. Multivalent-effect immobilization of reduced-dimensional perovskites for efficient and spectrally stable deep-blue light-emitting diodes. Nat. Nanotechnol. (2025).
https://doi.org/10.1038/s41565-024-01852-6